添加日期:2017年10月19日 阅读:2481
全球新药研发管线里程碑事件有哪些?本文就此进行简单的梳理。

目 录
一、批准与上市
1.Naldemedine-在美国上市
2.Abemaciclib-在美国上市
3.带状疱疹疫苗(Shingrix)-获加拿大批准
4.Ustekinumab-FDA批准扩大适应证人群
二、行政申报、审评、审批与资格认证
1.DAS181-获突破性疗法认证
2.Osimertinib-新适应证获FDA突破性疗法的认证
3.Gilteritinib-获FDA的突破性疗法认证
4.Uliprisnilacetate-NDA获受理
5.Tebipenem pivoxil-获QIDP认证
6.Pegfilgrastim生物类似药(Mylan&Biocon)-收到FDA的CRL
7.Voretigeneneparvovec-获FDA**委员会一致推荐
8.舒芬太尼舌下片(DSUVIA)-收到FDA的CRL
9.Apalutamide-提交NDA
10.Eryaspase-重新递交MAA
11.Rucaparib-向FDA提交sNDA
三、临床试验
1.PC-mAb启动Ⅱa期临床试验
2.Tinostamustine启动Ⅰ/Ⅱ期临床试验
四、商业行为
1.KVD-001-开发权转让
2.INB03-开发权转让
2017年10月9日至10月15日
一 批准与上市
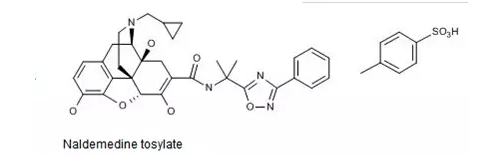
1.Naldemedine-在美国上市
根据Thomson Reuters Integrity报告,由日本盐野义制药株式会社开发的δ-阿片受体与μ-阿片受体双重拮抗剂Naldemedine 0.2mg片(商品名Symproic)于近日在美国上市。此前,Symproic于2017年3月获FDA批准用于治疗非癌性疼痛患者因使用阿片类镇痛药而引起的便秘,其MAA尚处于EMA的审查阶段。
盐野义制药在Symproic获FDA批准时发布的公告援引相关数据称,便秘是阿片类药物在其用药过程(包括用于治疗慢性非癌性疼痛)中所致的*常见的一种不良反应。非癌性疾病患者中,阿片类药物所致便秘的患病率约在40-50%之间。
Naldemedine药用其对甲苯磺酸盐,结构式如下图所示。
Thomson Reuters Integrity报告中收载了与Naldemedine有关的三组专利。分别是:
化合物专利:US8536192,其中国同族专利CN200680027118.7已获授权,将于2026年5月到期。
晶型专利:US9315512,其中国同族专利CN201180064907.9(原案)与CN201510332439.9(分案)均已获授权,同时将于2031年11月到期。
剂型/组合物专利:US2015216804,暂未进入中国。
2.Abemaciclib-在美国上市
根据Thomson Reuters Integrity报告,礼来公司开发、申报并成功获批的Pim-1激酶、CDK6与CDK4抑制剂Abemaciclib于近日在美国上市。此前,FDA于2017年9月28日批准Abemaciclib 50mg、100mg、150mg及200mg片(商品名:Verzenio)联合氟维司群用于治疗先前采用内分泌疗法但病情进展的HR-阳性/HER2-阴性的晚期或转移性乳腺癌患者,同时获批的适应证还有单独给药用于治疗先前采用内分泌疗法后病情进展的HR-阳性/HER-阴性的晚期或转移性乳腺癌患者以及先前采用化疗后病情进展的HR-阳性/HER2-阴性的转移性乳腺癌患者。Abemaciclib是FDA继palbociclib(商品名Ibrance,辉瑞公司)与ribociclib(商品名:Kisqali,诺华公司)之后批准的第个用于治疗乳腺癌的CDK抑制剂,其中的Ibrance于2015年2月获FDA批准,而2016年就实现了21.35亿元的年度销售额。
FDA在其批准Abemaciclib时发布的公告援引国家癌症研究院的数据称,2017年美国将新增25.271万名乳腺癌患者,另有4.061万名患者因乳腺癌死亡。约72%乳腺癌的肿瘤呈HR-阳性与HER2-阴性。
Abemaciclib药用其甲磺酸盐,结构式如下图所示。
中国专利CN200980151778.X审定/授权说明书的权利要求9以化学结构式的形式保护了Abemaciclib及其可药用盐,权利要求10将该发明所保护化合物的可药用盐限定为甲磺酸盐。
3.带状疱疹疫苗(Shingrix)-获加拿大批准
葛兰素史克公司于当地时间2017年10月13日发布公告称,加拿大卫生行政部门日前批准了该公司的带状疱疹疫苗(Shingrix)用于预防年龄≥50岁人群的带状疱疹病毒感染。Shingrix是一种非活亚单位疫苗,结合了一种抗原糖蛋白E和佐剂系统AS01B,该佐剂系统旨在产生强而持久的免疫反应,可帮助克服免疫力随年龄增长而下降。Shingrix开发用于50岁及以上人群,预防带状疱疹及其并发症。目前,该疫苗在美国、欧盟、加拿大、澳大利亚、日本的监管审查正在进行中。如果获批,该疫苗将通过2剂肌注(2针免疫程序:0月和2月)进行免疫接种。此前,瑞士银行预测,Shingrix上市后的年销售峰值将突破10亿美元大关。
在美国,GSK于2016年底向FDA提交了Shingrix的生物制品许可申请(BLA)。该BLA的提交,是基于一项全面的III期临床项目,该项目涉及18个国家(包括日本)超过37000名受试者,评估了Shingrix的有效性、安全性和/或免疫原性。其中,ZOE-50研究(NCT01165177)在16160例50岁及以上老年群体中开展,ZOE-70研究(NCT01165229)在14800例70岁以上老年群体中开展。数据显示,Shingrix不仅能够有效降低带状疱疹的发病率,而且能够降低带状疱疹后遗神经痛(PHN)的总体发病率,表明该疫苗针对已经感染带状疱疹的患者也具有一定的疗效。PHN是一种由带状疱疹引起的慢性疼痛,是感染带状疱疹后的典型症状。2017年9月中旬,FDA的疫苗及相关制品顾问委员会以全票通过的投票结果,表示认可Shngrix的有效性与安全性。
带状疱疹(shingles)是由水痘-带状疱疹病毒(VZV)引起的急性感染性皮肤病,典型表现为皮疹。由于病毒具有亲神经性,感染后可长期潜伏于脊髓神经后根神经节的神经元内,当抵抗力低下或劳累、感染、感冒时,病毒可再次生长繁殖,并沿神经纤维移至皮肤,使受侵犯的神经和皮肤产生强烈的炎症。皮疹一般有单侧性和按神经节段分布的特点,有集簇性的疱疹组成,并伴有疼痛;年龄愈大,神经痛愈重。该病好发于成人,发病率随年龄**而呈显着上升,并发症包括结疤、视力并发症、继发性感染、神经麻痹和带状疱疹后遗神经痛(PHN)。
据估计,在美国每年有100万例带状疱疹病例,该病发病率在北美、欧洲、亚太地区相似。老年人以及因病导致免疫力下降的人群,感染带状疱疹病毒的风险*高。来自许多国家的数据表明,有超过90%的成年人伴有带状疱疹风险,50岁以上人群感染带状疱疹风险显着升高。
4.Ustekinumab-FDA批准扩大适应证人群
强生公司于当地时间2017年10月13日发布消息称,FDA日前批准了该公司的Ustekinumab(商品名:Stelara)用于治疗年龄≥12岁且适用光疗或全身疗法的青少年斑块状银屑病患者。
Ustekinumab是一种人体白介素-12与-23拮抗剂,*早于2009年9月获FDA批准用于治疗年龄≥18岁且适用于光疗或全身疗法的成年斑块状银屑病患者。强生的公告指出,Ustekinumab因其两剂的起始剂量后每年只需给药4次,从而成为皮肤科医生与其患者的优选药物。
强生公司在其公告中还援引数据称,银屑病是一种慢性、自身免疫性的炎症疾病,可导致皮肤细胞的过度增殖。约750万美国人患有银屑病,其中80~90%为斑块状银屑病患者。此外,银屑病患者中,80%呈轻度至中度,而20%的患者呈中至重度的斑块状银屑病。青少年的斑块状银屑病多发于头皮与面部,从而对其情绪与社交产生严重影响。
Ustekinumab的Thomson Reuters报告中收载了14组与之相关的专利,其中*早的是US6902734,其中国同族专利CN01816961.9被驳回,但后续的分案申请200810100025.3已获授权,审定授权说明书的权利要求1保护了“一种分离的抗-IL-12抗体,其包含SEQIDNO:7所示氨基酸序列的重链可变区(VH)和SEQIDNO:8所示氨基酸序列的轻链可变区(VL),所述抗体为人抗体”,有效期截至2021年8月。
二 行政申报、审评、审批与资格认证
1.DAS181-获突破性疗法认证
总部设在美国圣地亚哥的Ansun BioPharma公司于当时时间2017年10月11日发布公告称,该公司开发用于治疗下呼吸道副禽流感病毒感染的新药DAS181被FDA认定为“突破性疗法”。
DAS181是利用大肠杆菌而制备得到的重组融合蛋白,其作用靶点是存在于人体呼吸道内的唾液酸受体,大部分呼吸道病毒感染宿主时均与此受体结合,而DAS181则通过裂解唾液酸受体而阻断呼吸道病毒对人体的感染。研究证实,DAS181对人体内四种主要的呼吸道病毒-流感病毒(IFV)、副流感病毒(PIV)、偏肺病毒(MPV)以及人肠道病毒-68(EV-68)均有抗病毒作用。DAS181独特的作用机制使得病毒无法依靠唾液酸受体进入宿主并产生耐药性,而临床前与临床研究中也未观察到耐药性或仅有极弱的耐药性。
Ansun BioPharma于*近完成了一项DAS181用于治疗免疫受损的住院患者PIV感染的2期临床试验,而其3期临床试验已处于筹备阶段。此外,在Ansun BioPharma与FDA的合作下,已有130名儿科与成人患者通过“同情用药”渠道接受了DAS181的治疗。
就其核心专利而言,Thomson ReutersIntegrity报告中所收录的与DAS181相关的*早专利是US8512710(优先权日期为2002年11月22日),该专利的中国同族专利(原案)CN200380107241.6已获授权,有效期截至2023年11月,另有分案申请CN200910180019.8被视为撤回。
2.Osimertinib-新适应证获FDA突破性疗法的认证
阿斯利康公司于当时时间2017年10月9日发布公告称,该公司开发并销售的第三代EGFR酪氨酸激酶抑制剂Osimertinib(Tagrisso)用于转移性EGFR突变-阳性非小细胞肺癌(NSCLC)的一线治疗的适应证获FDA的突破性疗法认证。作为此次突破性疗法认证之基础的Ⅲ期FLAURA试验以先前未接受过治疗的局部晚期或转移性EGFR突变阳性NSCLC患者为对象,比较了Tagrisso与标准酪氨酸激酶抑制剂(TKI)的有效性与安全性。结果发现,Tagrisso与标准TKI(厄洛替尼或吉非替尼)的中位无进展生存期分别为18.9个月与10.2个月。按照有无脑转移而事先划分的各亚组患者亦观察到有改善的现象。该临床试验中Tagrisso耐受性良好,其安全性特征与先前观察到的结果一致。
目前,已有包括美国、欧盟、日本与中国在内的50多个国家或地区批准Osimertinib每天给药一次的40mg与80mg片剂用于治疗EGFRT790M突变阳性晚期NSCLC。另有临床试验正在考察Osimertinib用于辅助治疗或其他治疗联合用药时的有效性与安全性。
Osimertinib药用其甲磺酸盐,结构式如下图所示。

中国专利CN201280033773.9审定/授权说明书的权利要求1即以化学名的形式保护了Osimertinib或其可药用盐,权利要求2保护了Osimertinib,而权利要求3则进一步保护了Osimertinib的甲磺酸盐。
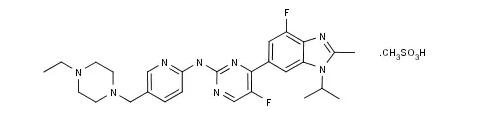
3.Gilteritinib-获FDA的突破性疗法认证
总部位于日本东西的安斯泰来制药于当地时间2017年10月11日发布公告称,由该公司开发用于治疗携带FLT3突变阳性的反复性或难治性急性髓样白血病(AML)的新药Gilteritinib被FDA认定为“突破性疗法”,从而为其加速临床开发并审批上市创造了有利的条件。
安斯泰来在其公告中援引美国癌症协会的数据称,美国每年约新增2.1万例AML病例,死亡1万例。约1/3的AML患者带有两种*常见的FLT3突变,即FLT3内部串联复制区与FLT3酪氨酸激酶结构区,而Gilteritinib对此两种蛋白区均有抑制作用。此外,Gilteritinib对据报道能引起治疗耐受的AXL也有抑制作用。
目前,安斯泰来公司正在进行4项Ⅲ期临床试验,评价Gilteritinib在多种AML患者中的有效性与安全,其中即包括了以反复性/难治性FLT3+AML患者为对象的ADMIRAL试验。
Gilteritinib的结构式如下图所示。
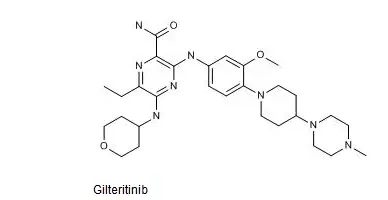
中国专利CN201080020181.4审定授权说明书的权利要求14以化学名的形式保护了Gilteritinib或其可药用盐,该专利有效期截至2030年5月。
4.Uliprisnil acetate-NDA获受理
总部位于都柏林的Allergan制药公司于当地时间2017年10月10日发布公告称,FDA于近日受理了该公司先前提交的Uliprisnil acetate用于治疗子宫平滑肌瘤的NDA,如果成功通过审查,Uliprisnil acetate则将是**用于治疗子宫平滑肌瘤的口服药物。
Allergan公司在其公告中援引卫生保健与质量管理局(AHRQ)的分析结果称,在美国约有2600名15~50岁之间的女性患有子宫肌瘤,其中至少一半的患者日常生活因该疾病的症状而受到影响。子宫肌瘤的主要症状有月经期失血过多、月经周期**、月经周期不规则、贫血、骨盆疼痛、骨盆压力与泌尿道症状等。
Uliprisnil acetate是一种选择性孕激素受体调节剂(SPRM),其直接作用于子宫内膜、子宫平滑肌瘤与脑下垂体内的孕激素受体。北美地区两项纳入500余名育龄期女性的Ⅲ期临床试验(VenusⅠ与VENUSⅡ),以及欧洲地区纳入1000余名子宫肌瘤女性的4项多中心Ⅲ期临床试验对Uliprisnilacetate的安全性与有效性进行了系统的评价。
此前,Uliprisnil acetate已在欧洲与加拿大上市,商品名分别为Esmya与Fibristal,分别由Gedeon Richter公司与Allergan公司销售。
Uliprisnil acetate结构式如下图所示。

Thomson Reuters Integrity报告中收载的Uliprisnil acetate*早的专利是US4954490及其同族专利,然而该专利未进入中国。
5.Tebipenem pivoxil-获QIDP认证
总部位于美国马萨诸塞州剑桥市的Spero Therapeutics公司于当地时间2017年发布公告称,日前该公司与日本明治制果制药株式会社就SPR994(Tebipenem pivoxil)的开发达成了排他性的合格协议,Spero Therapeutics公司同时宣布,SPR994还被FDA认定为QIDP,即“合格感染疾病产品”,其适应证包括复杂性泌尿道感染(CUTI)、糖尿病足(DFI)与社区获得性细菌性肺炎(CABP)。
SPR994是由日本明治制果公司开发的一种广谱口服碳青霉烯类抗菌药,用于治疗多药耐药性革兰阴性细菌所致的感染,其中包括对氟喹诺酮耐药的感染性疾病。该药已在日本(2009年,商品名Orapenem)与多个亚洲国家上市销售。SPR994的临床与药理研究中,共有约1200名万年与儿童患者接受了该药的治疗。
SPR994的化学结构式如下图所示。
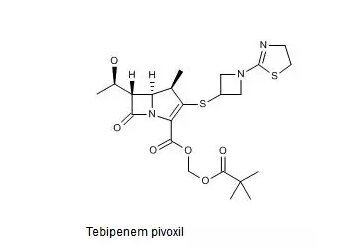
Thomson Reuters Integrity报告中收载与SPR994相关的*早的专利是US4644061及其同族专利,然而该专利未进入中国。
6.Pegfilgrastim生物类似药(Mylan & Biocon)-收到FDA的CRL
总部位于印度新德里的Biocon制药公司于当地时间2017年10月10日发布公告称,FDA日前就该公司与Mylan制药公司联合开发并申报的Pegfilgrastim生物类似药(MYL-1401H,)发布了完整回复涵(CRL),暂时不批准该生物类似物。
Biocon制药公司与Mylan制药公司正在全球范围内就6个生物类似物的开发进行合作,MYL-1401H就是其中之一。Biocon公司的发言人表示,此次CRL将不会影响MYL-1401H在美国的上市计划,该公司将与FDA密切沟通,以及时解决CRL中所提出的问题。
7.Voretigeneneparvovec-获FDA**委员会一致推荐
总部位于美国费城的Spark Therapeutics公司于当地时间2017年10月12日发布公告称,FDA的组织与基因疗法顾问委员会在日前的例行会议上以16:0的投票结果通过决议,表示该公司开发并申报用于治疗双等位基因RPE65介导的遗传性视网膜疾病所致失明的一次性基因疗法LUXTURNA(Voretigeneneparvovec)效益大于风险,从而使得Voretigeneneparvovec有望成为继诺华CAR-T之后的第二个获FDA批准的基因疗法。
Luxturna疗法由费城Spark Therapeutics公司研制,旨在治疗RPE65基因拷贝数变异引起的疾病。这种变异会影响眼睛对光的反应能力,*终导致视网膜感光系统失能。
该疗法应用装载有RPE65基因的正常拷贝的病毒,将病毒注射到患者眼睛中并使其中基因表达,提供RPE65编码的正常蛋白。
在一项涉及31人的随机对照试验中,Luxturna在接受治疗的患者身上都表现出一定的疗效,SparkTherapeutics将继续对这一疗法进行研究。
Thomson Reuters Integrity报告中只收载了3组与Voretigeneneparvovec有关的专利,均被标注为基础专利。按照优先日期从早到晚依次是US2004022766(未进入中国)、US2007077228(未进入中国,与上一专利互为同族专利)、CN201180016616.2(中国专利,复审中)。
8.舒芬太尼舌下片(DSUVIA)-收到FDA的CRL
总部位于美国加利福尼亚州的AcelRx Pharmaceuticals于当地时间2017年10月12日发布公告称,FDA日前就该公司所提交的DSUVIA(舒芬太尼舌下片,30mcg)NDA发布了完整回复涵(CRL),表示暂时不批准该NDA。
阿片类镇痛药舒芬太尼(药用其枸橼酸盐)临床上用于静脉及硬膜麻醉与镇痛,因为静脉给药后的作用持续**从而限制了其临床应用。AcelRx Pharmaceuticals开发的舒芬太尼30mcg舌下片就旨在克服静脉注射剂作用**的缺点,该药的MAA(名称ARX-04)亦正在接受EMA的审查。
AcelRx Pharmaceuticals相关负责人表示,虽然FDA暂时不批准DSUVIA,但是该CRL中所提出的问题均未涉及实质性缺陷。AceRx Pharmaceuticals将与FDA保持密切沟通,以期继续推动DSUVIA在美国的上市。
根据Thomson Reuters Integrity报告,枸橼酸舒芬太尼注射剂于1982年即已上市,因此其化合物专利早已到期。根据AcelRx Pharmaceuticals于2012年8月29日发布的公告,该公司针对DSUVIA进行了一系列的专利而已,其中*近的US8252328与US8252329均涉及舒芬太尼。此外,espacenet数据库中以AcelRx进行检索,共检出22项专利,由于涉及制剂技术,本文不对其进行扩展论述。
9.Apalutamide-提交NDA
强生制药于美国当地时间2017年10月11日发布公告称,该公司日前向FDA提交了新一代口服雄激素受体(AR)抑制剂apalutamide用于治疗男性非转移性去势抵抗性前列腺癌(非转移性CRPC)的NDA,目前FDA暂未批准任何药物用于治疗非转移性CRPC。
强生公司在其公告中援引美国癌症协会的数据称,2017年美国有超过16.1万名男性被确诊为前列腺癌。接受雄激素剥夺治疗(ADT)的非转移性前列腺癌患者*终会对ADT产生耐药性,从而发展为CRPC。有数据显示,10%~20%的前列腺癌确诊患者会在5年内进展为CRPC。
强生公司的公告还指出,此次申报以关键性Ⅲ期临床试验ARN-509-003(SPARTAN)的数据为基础,该项试验以连续接受ADT治疗后前列腺特异抗原(PSA)仍然迅速上升的非转移性CRPC男性患者为对象,并以安慰剂为对象,评价了apalutamide的有效性与安全性。强生公司表示,将会在将来的医学会议上公布该项试验的结果。
Apalutamide的结构式如下图所示。
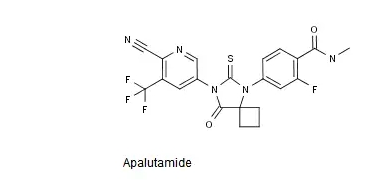
中国专利CN200780019654.7审定/授权说明书的权利要求6以化学结构式的形式单独保护了Apalutamide或其可药用盐,该专利将于2027年3月失效。
10.Eryaspase-重新递交MAA
总部位于法国里昂的ErytechPharma公司于当地时间2017年10月10日发布公告称,该公司日前向EMA重新递交Eryaspase(商品名:Graspa)用于治疗反复性或难治性急性成淋巴细胞白血病(ALL)的MAA
Eryaspase是由ErytechPharma开发,将L-天门冬酰胺酶包囊于人红细胞内而制得的产品。2015年9月,该公司向欧盟EMA递交了Eryaspase用于治疗ALL的MAA,但却于去年11月撤回了此MAA,以争取足够的时间提供CHMP所要求的其他信息。此次重新递交的申报资料中包括了以儿童与成年ALL患者为对象的Ⅱ/Ⅲ期临床试验GRASPALL2009-06(Clinical Trials.gov识别号:NCT01518517)以及为了回答CHMP的问题而需要的其他数据。GRASPALL2009-06试验结果显示,与原态L-天门冬酰胺酶相比,eryaspase联合化疗的治疗方案对反复性或难治性ALL患者显示出良好的有效性与安全性特征。Eryaspase治疗组患者L-天门冬酰胺酶活性持续时间长度几乎是原态L-天门冬酰胺酶治疗组患者的两倍。该项试验中,Eryaspase亦显示出良好的安全性特征,无患者在接受eryaspase治疗的过程中出现过敏反应,而原态L-天门冬酰胺酶治疗组内过敏反应的发生率为48%。eryaspase治疗组内患者在诱导期内的整体完全康复率高于原态L-天门冬酰胺酶治疗组,而药物相关性不良反应的发病率却低于后者。
该药尚未在任何国家获准上市。
Thomson Reuters Integrity报告援引美国癌症协会的数据称,2017年美国将新增6.213例白血病病例,每种白血病新增病例如下所示:
急性淋巴细胞白血病(ALL):5970例;
慢性淋巴细胞白血病(CLL):2.011万例;
急性髓性白血病(AML):2.318万例;
慢性髓性白血病(CML):8950例;
其他类型的血病:5720例。
该药的Thomson Reuters Integrity报告中只收载了3组与之相关的专利:
US8617840,中国同族专利CN200580029814.7审定/授权说明书的主权项保护了“制备包含活性成分的红细胞的裂解/再封方法”,该专利尚在有效期内,有效期截至2025年8月;
US8974802,中国同族专利CN200880126046.0申请/公开说明书的主权项要求保护“用于治疗胰腺癌的包封天冬酰胺酶之红细胞的混悬剂”,但该专利已被驳回,其分案申请CN201610012474.7尚在审查阶段;
WO2017114966要求保护采用蛋氨酸酶与天冬酰胺酶治疗癌症的方法,尚未进入中国。
11.Rucaparib-向FDA提交sNDA
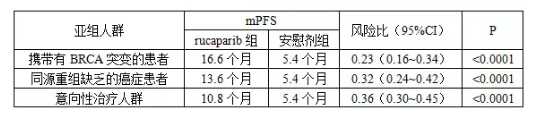
总部位于美国科罗拉多州的Clovis Oncology于当地时间2017年10月9日发布公告称,该公司日前向FDA递交了Rucaparib用于先前采用铂类化疗后病情完全缓解(CR)或部分缓解(PR)的反复性卵巢上皮癌、输卵管癌或原发性腹膜癌患者维持治疗的sNDA。作为此次sNDA申报基础的Ⅲ期双盲、安慰剂对照临床试验(ARIEL3试验,Clinical Trials . gov识别号:NCT01968213)共招募了564名铂敏感卵巢癌、输卵管癌或原发性卵巢癌女性患者。该项临床试验的主要有效性终点分析由从高到低的3个亚组人群构成,根据发表于《柳叶刀》的研究结果(Coleman RL, et al. ARIEL3 investigators. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017 Sep 12. pii: S0140-6736(17)32440-6.),不同人群的中位无进展生存期(mPFS)结果如下表所示。
安全性分析人群中,rucaparib组与安慰剂组内3级或更高级别治疗相关性不良反应的发病率依次为56%与28%,*常见的不良反应是贫血或血红蛋白浓度下降(19% vs 1%)以及丙氨酸或天冬氨酸转氨酶水平上升(10% vs 0)。
Rucaparib是一种多聚ADP-核糖聚合酶(PARP)抑制剂,通过作用于PARP-1、PARP-2和PARP-3修复DNA起作用。2016年12月,FDA通过快速审批程度批准了Rucaparib 200mg、300mg与250mg片(商品名:Rubraca)作为单一疗法用于治疗接受过两次及以上化疗的BRCA突变的晚期卵巢癌患者。
Clovis Oncology在其公告中援引美国癌症协会的数据称,2017年美国将新增2.24万名卵巢癌病例。卵巢癌在其发病早期并无清晰可辨的症状,而80~85%的卵巢癌患者在其确诊之前肿瘤即已扩散至身体的其他部位,并可接受治疗。卵巢癌所致的死亡病例数在癌症所致的死亡病例中排名第五,且高于其他妇科生殖系统癌症。
Rucaparib药用其右旋樟脑磺酸盐(camsylate),结构如下图所示。
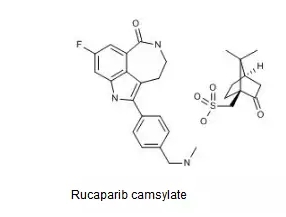
中国专利CN00804589.5审定/授权说明书的权利要求3以化学结构式的形式保护了包括Rucaparib在内的多个三环类PARP抑制剂及其可药用盐,该专利有效期截至2030年1月。
中国专利CN201180009237.0审定/授权说明书的权利要求1以化学名的形式保护了Rucaparib樟脑磺酸盐,权利要求3与4分别对所述樟脑磺酸盐的构型予以了限定,该专利的有效期截至2031年2月。
三 临床试验
1.PC-mAb启动Ⅱa期临床试验
总部位于瑞斯德哥尔摩的Athera Biotechnologies AB公司于当地时间2017年10月11日发布公告称,该公司启动了一项评价PC-mAb对动脉血管炎症的治疗作用的前瞻性、双盲、随机化、安慰剂对照的多中心临床试验。该项试验的受试者人群由脂蛋白a(Lp(a))水平升高的患者构成,所述的脂蛋白a对磷脂酰胆碱(PC)有较强的携带能力,而PC则是PC-mAb促炎症反应的治疗靶点。
2.Tinostamustine启动Ⅰ/Ⅱ期临床试验
总部位于瑞士的MundipharmaEDO公司于当地时间2017年10月10日发布公告称,该公司近日在美国启动了tinostamustine(EDO-S101)治疗晚期实体瘤患者的Ⅰ/Ⅱ期临床试验。该项试验Ⅰ期部分的主要目的是评价tinostamustine单独给药时的安全性、耐受性、*大耐受剂量并为Ⅱ期临床试验筛选剂量。除了以评价受试者药代动力学特征作为其次要目的,该项试验还将探索性考察带有抗肿瘤活性的肿瘤样本中基因表达的变化程度,而其Ⅱ期部分的受试者则将包括小细胞肺癌、软组织肉瘤、非KIT胃肠道间瘤、三阴性乳腺癌与鸟巢癌等具体实体瘤的患者。
Tinostamustine是一种脱乙酰酶抑制剂类烷化剂,其化学结构式如下图所示。
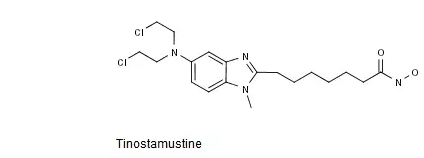
中国专利CN201080002890.X审定授权说明书的权利要求3以化学结构式的形式单独保护了Tinostamustine或其可药用盐,该专利有效期截至2030年1月。
四 商业行为
1.KVD-001-开发权转让
总部设在美国马萨诸塞州剑桥市的KalVista公司于当地时间2017年10月10日发布公告,默克Merck & Co(NYSE:MRK)已经出资910万美元(8.50美元/股)取得该公司9.9%的所有权。
此外,默克还将支付KalVista 3700万美元预付款获得收购其糖尿病性黄斑水肿候选药的期权,两家公司将合作开发该药物。另外,默克还获得未来其他口服糖尿病性黄斑水肿化合物的类似期权。KalVista还有可能获得*高7.15亿美元的额外支付款,以及基于净销售额的分层专利权许可费。
KalVista计划开展糖尿病性黄斑水肿治疗药物KVD001的二期试验。糖尿病性黄斑水肿是一种糖尿病并发症,可能导致视力受损或失明。KalVista股价周一收盘报7.35美元,过去三个月上涨0.3%,同期标普500指数(SPX)上涨4.8%。Merck股价盘前上涨0.5%。
KVD-001是一种血浆激肽释放酶抑制剂。暂时未查到与其结构式以及专利有关的信息。
2.INB03-开发权转让
总部位于美国加利福尼亚州的 INmune Bio 公司于当地时间2017年10月10日发布公告称,该公司日前从 Xencor Inc. 公司获得了**免疫检查点抑制剂INB03的开发权。公告指出,大部分癌症患者**免疫系统的髓源抑制细胞(MDSC)水平均有所上升。血液或肿瘤微环境内的MDSC水平能用于预测疾病的严重程度,癌症的致死风险以及包括已有检查站抑制剂在内的其他免疫疗法治疗失败的可能性。MDSC所分泌的免疫抑制性细胞因子能使肿瘤免受人体免疫系统的攻击,而抑制MDSC则可有效地推动癌症治疗的发展.
责任编辑:成浩 WWW.1168.TV 2017-10-19 11:40:36
文章来源:
1.凡本网注明“来源:1168医药招商网”的所有作品,均为广州金孚互联网科技有限公司-1168医药招商网合法拥有版权或有权使用的作品,未经本网授权不得转载、摘编或利用其它方式使用上述作品。已经本网授权使用作品的,应在授权范围内使用,并注明“来源:1168医药招商网http://www.1168.tv”。违反上述声明者,本网将追究其相关法律责任。
2.本网转载并注明自其它来源(非1168医药招商网)的作品,目的在于传递更多信息,并不代表本网赞同其观点或和对其真实性负责,不承担此类作品侵权行为的直接责任及连带责任。
3.其他媒体、网站或个人从本网转载时,必须保留本网注明的作品第一来源,并自负版权等法律责任。
4.如涉及作品内容、版权等问题,请在作品发表之日起一周内与本网联系,否则视为放弃相关权利。联系邮箱:1753418380@qq.com。

【适用范围】用于缓解颈、肩、腰、腿及闭合性软组织疼痛、肿胀等不适症状人群的物理冷敷。【使用方法】外用。将本品适量直接涂抹于不适部位,轻轻按摩2-3分钟,每日2-3次。

【适用范围】用于缓解颈、肩、腰、腿及闭合性软组织疼痛、肿胀等不适症状人群的物理冷敷。【使用方法】外用。将本品适量直接涂抹于不适部位,轻轻按摩2-3分钟,每日2-3次。
 粤公网安备 44011102000390号
粤公网安备 44011102000390号